
9 Must-knows for US biotechs looking to enter the EU Market
26 April 2023
Indzine
Late-stage American biopharmaceutical companies in late-stage development for their product are logically interested in the European market, and we clearly see a trend of companies looking to expand in Europe by themselves rather than partnering with an establishment based there.
If we take a quick look at the map representing market share of new drugs launched between 2015 and 2020, it is quite easy to understand that Europe certainly is a market of interest.
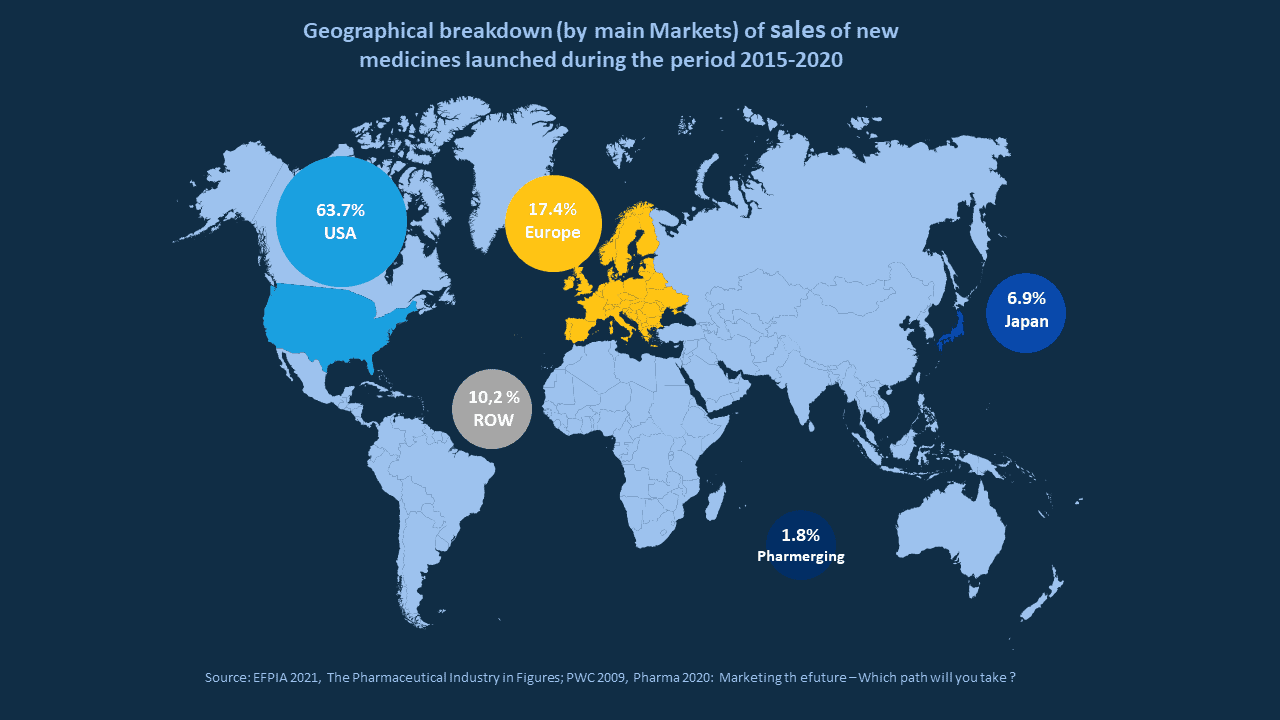
If you want to ensure that your registration & launch strategy in Europe are as successful as possible, it's important to be aware of the regulatory affairs aspects and how they can impact your strategy.
We are an expanding regulatory affairs consultancy specializing in the European market and our capabilities include supporting pharmaceutical companies in their ability to develop and offer healthcare products to patients, including innovative treatment.
Today we are happy to share with you 9 tips for US-based companies looking to register and launch their product on the European market.
1. SME Status / SME Representation

One of the great incentives for companies developing pharmaceuticals in Europe is the Small and Medium-sized Enterprise (SME) Status delivered by the European Medicines Agency (EMA).
This EU incentives offered by the Agency includes Regulatory, Administrative and Procedural assistance from the EMA, Fee reductions or exemptions, Certification of quality/non-clinical data for Advanced Therapy Medicinal Products (ATMPs) and tax credits, research grants.
SME status itself consequently can incur lower costs for dossier submissions.
European consultancy companies usually provide SME Status representation for their clients who do not have an affiliate in EU and can be eligible to SME status. Consultancies can act as their sponsor for their SA, PIP, ODD, MA.
SA: Scientific Advice
PIP: Pediatric Investigation Plan
ODD: Orphan Drug Designation
MA: Market Authorization
2. ATMPS incentives
Developers of ATMPs can obtain reductions in the fees payable to EMA of:
- 65% fee reduction for a request for scientific advice for ATMPs (90% for SMEs);
- 90% fee reduction for the certification procedure.
You can find all the incentives available for advanced-therapy developers on EMA website : https://www.ema.europa.eu/en/human-regulatory/research-development/advanced-therapies/support-advanced-therapy-developers
3. EMA and EU national health agencies vs FDA

As regulatory requirements become increasingly harmonized across the globe, the development and marketing of pharmaceutical products worldwide are also becoming more streamlined. However, global regulations are not one-size-fits-all, and sponsors aiming to market their products in multiple regions should be aware of the current standards and processes they may encounter during the development process.
The FDA governs the drug and biologic approval process in the United States, while in Europe, each country has its own health national agency, and the EMA serves the European Union (EU) plus Iceland, Norway, and Liechtenstein.
The drug approval process represents one of the most obvious differences between US and EU agencies. The FDA oversees all drug approvals in the US via New Drug Applications (NDAs), with approvals for biologic products being approved via Biologics License Applications (BLAs).
In contrast, in Europe, there are multiple agencies and the 4 available approval pathways for pharmaceuticals can vary depending on your medicinal product and its current registration status. The 4 approval pathways are:
- Centralized
- Decentralized
- Mutual recognition
- National
While some products have specific requirements dictating which of these pathways is to be followed, in other cases sponsors should think carefully and select the approval procedures most suitable for their products. Similarly, there are differences between the scientific advice procedures, clinical trial procedures, and agency meetings.
Again, Regulatory affairs consultancies can help you create a time- and cost-efficient development strategy that enables you to attain approval in both the US and the EU.
4. CTIS
The Clinical Trials Information System (CTIS) is an information technology tool whose goes live on 31 January 2022.
During the first year after CTIS go-live, sponsors will be able to choose whether to apply for a new clinical trial application (CTA) under the regime of the Clinical Trial Directive (CTD: Directive 2001/20/EC) including using EudraCT or to apply under the new legislation, the Clinical Trial Regulation (CTR: Regulation (EU) No 536/2014) using CTIS. Both options will be possible, and sponsors will be able to choose for themselves which system to use.
From 31 January 2023 all new CTAs must be submitted under the new legislation (CTR) using CTIS.
CTIS will facilitate:
- Authorization of clinical trials in up to 30 EEA countries with a single application
- Involvement of trial participants by allowing easy expansion of trials to other EU/EEA countries
- Collaboration across borders for better results and knowledge sharing
- Clinical research investment, ensuring the EU/EEA remains an attractive location for clinical research
- Clinical trial publication requirements, for publication to occur with no additional effort
5. Early Access Programs

For certain rare, serious, or disabling diseases, the exceptional use of drugs before obtaining their Marketing Authorization (MA) or their coverage under common law may be authorized by some competent authorities.
Patients can benefit from the treatment ahead of the first European marketing authorization. Indeed, health products can reach market earlier, preparing for future commercial launch:
- Allowing continuous access to the drug for patients
- Building the company image before commercial launch
- Generate turnover before being approved
- Setting the scene for future market access of commercial product
👉 How to take advantage of early access programs for your innovative treatments in France?
6. Global and Local Specificities

Pharma companies may have several options to file a marketing authorization application for a new drug in the EU. One of these options, and if the product is eligible to this procedure, is the EMA centralized route which lead to a single marketing authorization in all EU countries. This marketing authorization is granted by the European commission following the scientific assessment of the application by the relevant committees of the EMA.
Once the pan-European Marketing authorization is granted, a large number of additional national regulatory activities in the EU member states are still necessary prior to the commercial launch of the drug in the concerned countries.
EU market:
30 countries (including France, Germany, Italy, Spain & Sweden) and 25 different languages.
Many different Health competent authorities and local requirements in these 30 member states.
The situation is even more complex for start-up companies, or when they are not yet based or established in the EU. The difficulties are to be compliant with all local regulations of these European countries. Hence the importance of implementing a regulatory strategy matching with the marketing expectations, taking into consideration the regulatory constraints as well as planning future geographical roll-out in additional international (non-EU) countries.
Therefore, a close coordination of all the stakeholders is required, and of course, without having a good understanding of the European regulatory landscape and appropriate representatives at each country level, it can be extremely difficult to execute the commercialization plan.
Furthermore, the language barrier is an additional burden, as part of the activities must be completed only in the local language of each country.
👉 How to accelerate your product launch in Europe
7. Promotional Activities - Responsible Persons
Once the drug has obtained a marketing authorization, promotional and educational activities (including materials) play a key part in the biotech and large pharma companies’ commercial strategy and as they now look to planning the launch of their new drugs in Europe.
However, such promotion must be compliant with pharmaceutical advertising laws imposed by European and/or national authorities.
👉 Anticipate and strategize promotional materials compliance in Europe

8. Establishing a “European Hub » location - Zug (Switzerland)
Zug is one of the preferred locations for US biotech looking to set up an affiliate in Europe. The city in northern Switzerland has become known as the Crypto Valley because of its close ties to the world of cryptocurrencies and blockchain technology, and it is now attracting more and more companies involved in what promises to be the next big thing: biomedicine.
Zug is one of two places in Switzerland to have opened a life-sciences cluster to cater specifically for the nascent industry and help companies set up shop as well as taking advantage of the local taxes systems.
9. Market Access specificities: Price Negotiation
The European Union provides a common framework for market access to ensure safe, effective, and high-quality medicines. The EMA is responsible for authorizing medicines in the European Union.
However, each country within the EEA has its own national health regulation with disparate Health Technology Assessment (HTAs) systems and processes. This means that every country has its own legislation and criteria in terms of price and reimbursement of a medicinal product.
The objectives of HTA bodies and payers are different from the health agencies which mainly assess the quality and benefit/risk ratio of a drug. HTAs bodies demand compelling evidence about how new medicines improve the standard of care and public health. It is sometime very difficult to negotiate fair price in Europe due to late consideration in the clinical trials of this added value of the drug versus the standard of care. In addition, the decentralized approach of the HTA bodies often results in different outcomes in EU countries. So, a careful Market Access strategy should be implemented at a very early stage in your European development and registration strategy.
We have seen a very good news recently from EMA as the Regulation on Health Technology Assessment (HTA) has been adopted.
This is a very important file that will contribute to the objectives of the Pharmaceutical Strategy for Europe in terms of supporting innovation, addressing unmet medical needs, and facilitating patient access to innovative medicines.
We hope that you’ve enjoyed this article, if you are a pharmaceutical company with a first important innovative product, going through the different stages of development or reaching the registration phase and looking for European regulatory requirements support, feel free to contact us!
Indzine
Did you like this article? Share on social networks:
